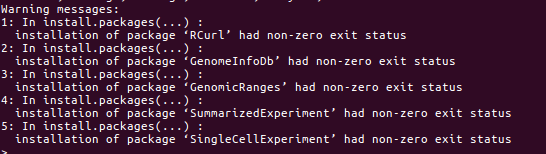
when I tried to install R packages “SingleCellExperiment” following this guide.
I came across the following errors.
when I tried to install the first package alone, I encountered another error
Error: package or namespace load failed for ‘RCurl’ in dyn.load(file, DLLpath = DLLpath, ...):
unable to load shared object '/home1/username/R/x86_64-pc-linux-gnu-library/4.2/00LOCK-RCurl/00new/RCurl/libs/RCurl.so':
libicui18n.so.70: cannot open shared object file: No such file or directory
Error: loading failed
Execution halted
ERROR: loading failed
P.S.: I also came across installation problems when installing Rfast package with the following error info
install.packages("Rfast")
Warning messages:
1: In install.packages("Rfast") :
installation of package ‘RcppGSL’ had non-zero exit status
2: In install.packages("Rfast") :
installation of package ‘RcppZiggurat’ had non-zero exit status
3: In install.packages("Rfast") :
installation of package ‘Rfast’ had non-zero exit status
when I tried to install RcppGSL, the following error msg occured
Error: package or namespace load failed for ‘RcppGSL’ in dyn.load(file, DLLpath = DLLpath, ...):
unable to load shared object '/home1/jiaweih/R/x86_64-pc-linux-gnu-library/4.2/00LOCK-RcppGSL/00new/RcppGSL/libs/RcppGSL.so':
libgsl.so.25: cannot open shared object file: No such file or directory
Error: loading failed
Execution halted
ERROR: loading failed
However I can install Rfast through a conda command
`conda install -c conda-forge r-rfast`
I was wondering what is the right way to install R packages,
install conda and create a R environment and install packages by ourselves
module load R in the shell and install.package() in R
Either way works, but there’s a potential tradeoff between ease-of-use and performance. With Conda, you’re creating your own software environment (independent of modules) and Conda will manage system and library dependencies. Using the modules, R packages are built from source and some R packages have system and library dependencies that need to be made available to the build process (sometimes this means just loading a module but sometimes this means specifying configure variables or even modifying a Makefile). Conda is downloading generic Linux binaries for these packages and dependencies, so these versions may run slower compared to the ones that are built from source using the modules. But it depends on the specific compiler options, packages, and functions that are used. Overall it’s easier to use a Conda environment for package management. But if you need to maximize performance, then using the modules and building from source is the better option.
I ran into problems both ways. When I installed through conda, I installed the Rfast but cannot install “SingleCellExperiment”, when I chose module installation. I cannot install both packages. Could you take a look and see how to solve the installation error above? (I prefer the module installation as you just pointed out).
With the modules, there are different steps for each package. For RCurl, I can install with the curl and libxml2 modules loaded. Then SingleCellExperiment installs like normal. For RcppGSL, I can install with the gsl module loaded. For RcppZiggurat, create or modify ~/.R/Makevars to include the following lines:
P.S.: I am really interested about how you find and know installing these packages with additional modules loaded ( and also modify Makevars file, I don’t even know what this file is used for). Could you share your experience with us so that we can know how to search for future issues.
It’s mostly working backwards from the specific error message. With an error like libgsl.so not found, I find out what software package provides that library file and then load the corresponding module and try installing the R package again. In this case, the build process also requires the development header files, so then I specify where to find the header files using the ~/.R/Makevars file (the CPPFLAGS variable). All R packages that compile objects use a general configure/make build process, but unfortunately they can be set up differently so there’s a different process to follow depending on the specific R package.
First load modules from HPC
module load openblas/0.3.18 r/4.1.2 gsl/2.7 curl/7.79.0
Install the software with the following command:
remotes::install_github(‘Jiaxin-Fan/MuSiC2’)
And I got the error message
Installing package into ‘/home1/jiaweih/R/x86_64-pc-linux-gnu-library/4.1’
(as ‘lib’ is unspecified)
ERROR: dependency ‘MuSiC’ is not available for package ‘MuSiC2’
removing ‘/home1/jiaweih/R/x86_64-pc-linux-gnu-library/4.1/MuSiC2’
I got the following error
Installing package into ‘/home1/jiaweih/R/x86_64-pc-linux-gnu-library/4.1’
(as ‘lib’ is unspecified)
ERROR: dependencies ‘TOAST’, ‘MCMCpack’ are not available for package ‘MuSiC’
removing ‘/home1/jiaweih/R/x86_64-pc-linux-gnu-library/4.1/MuSiC’
Warning messages:
1: package ‘TOAST’ is not available for this version of R
when I try to install ‘MCMCpack’, it still gives me an error. I don’t know what to do.