After the maintenance of the discovery cluster, when I use salloc to request a core to run my code, errors pops out like this. However, this does not happen before the maintence. Does anyone know why this happens?
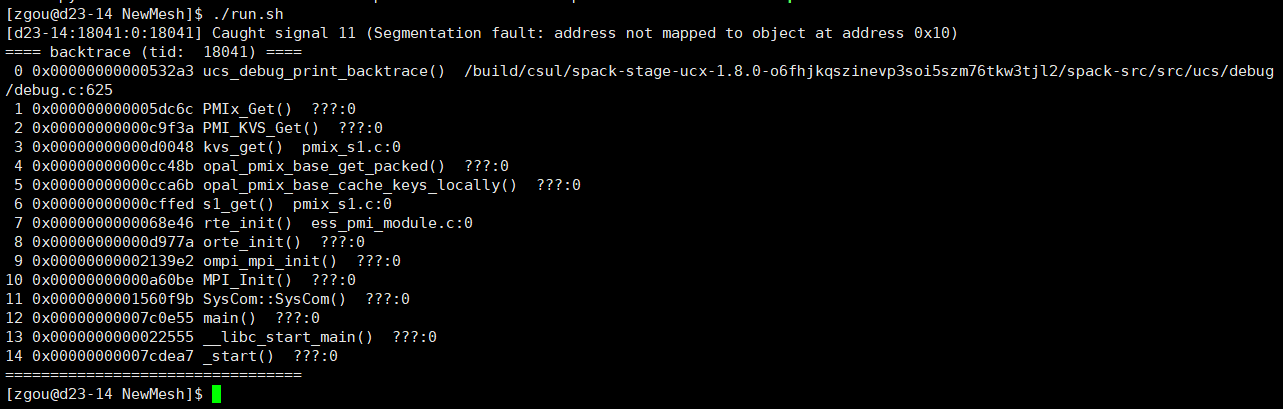
@zgou What modules did you have loaded? What does your shell script do? Could you share if possible?
I load usc module. And the shell script looks like this:
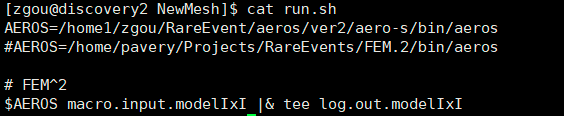
The AEROS is a software I self-compiled and installed in my folder. In fact, I can submit a slrum job to run AEROS with 6 cores. But I cannot do interactive jobs with salloc command to run AEROS. The slurm script looks like this:
#!/bin/bash
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=6
#SBATCH --time=1:00:00
#SBATCH --mem=5000MB
cd /home1/zgou/RareEvent/simulation/Hexbar.ymtt/NewMesh/
module load gcc/8.3.0 openmpi/4.0.2 pmix/3.1.3
export OMP_NUM_THREADS=6
./run.sh
And AEROS was compiled with openmpi/4.0.2? I see you’re only requesting 1 CPU for the interactive job. Try salloc --cpus-per-task=6 and see if it works then. There was a change to salloc with our recent Slurm update, so this issue is perhaps related to that.
Yes, AEROS is compiled with openmpi/4.0.2. Just now I used salloc to request 6 cpus to run this but result in the same error. I didn’t encounter this error before the maintenance.
Okay, I was able to reproduce this, but running the command with srun solved it. Try running srun ./run.sh.
It’s not working in my case. As the picture shows below, I requested 6 CPUS and use “srun ./run.sh”, then a new error shows “exec format error”. But when I exit the interactive mode and do “./run.sh” on the login node, it works although I am not supposed to do this on the login node. However, it does prove that there is no error in the exec format.
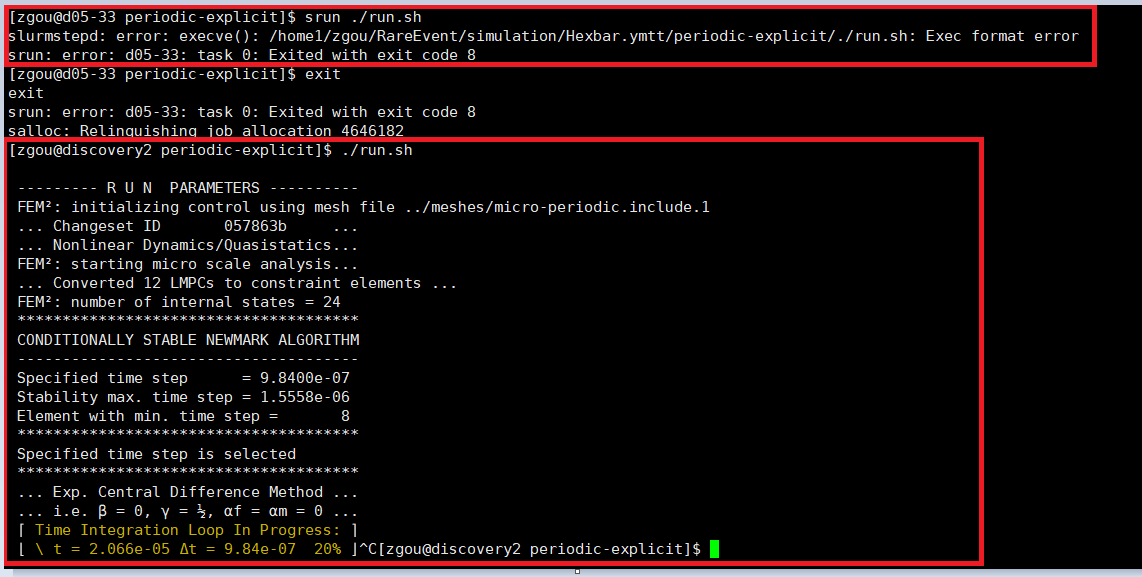
Ah, right. You’ll need to add #!/bin/bash to the top of your shell script.
Cool, it works! Thank you so much.