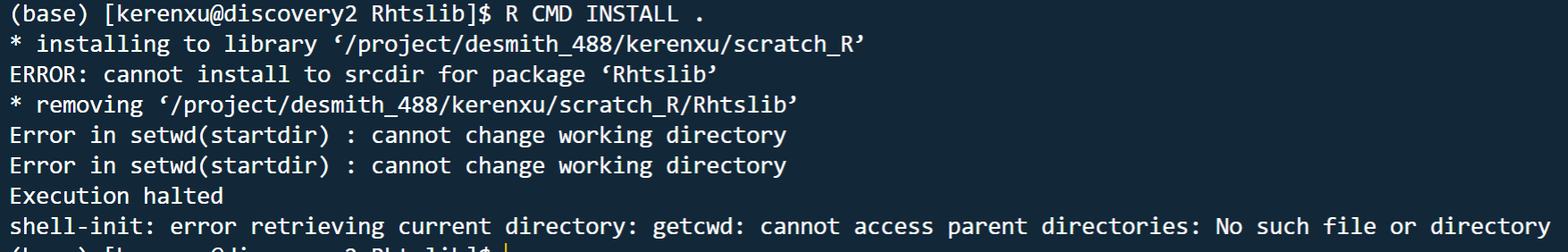
I was trying to install minfi package, but I kept seeing error messages. Similar messages occur when I was trying to install other bioconductor packages. I am using R 4.0.0 module. zlib.h seems to be missing. So several bioconductor packages were not installed, such as minfi, Rhtslib, GenomicAlighments, et al.
Below is the error message:
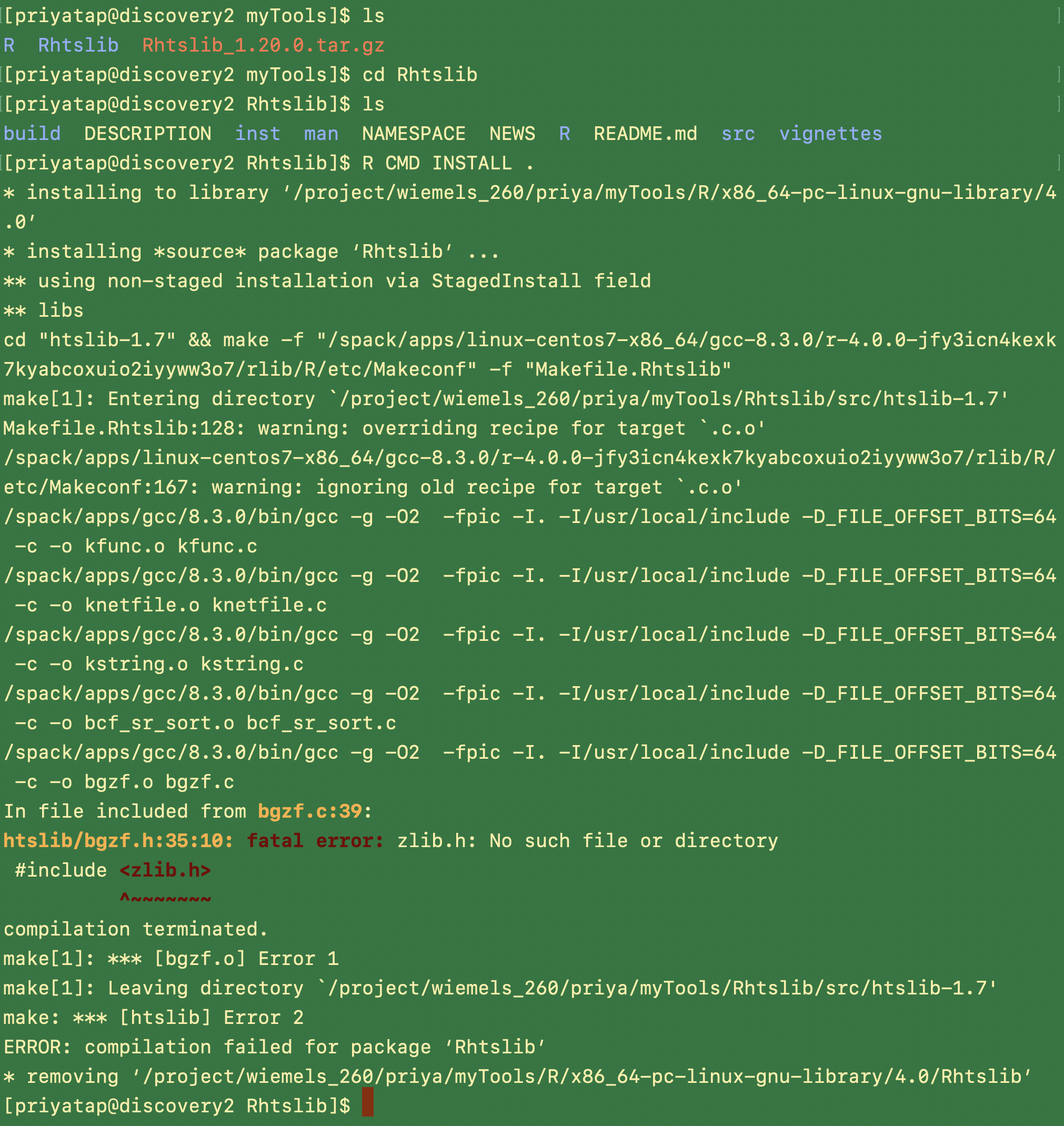
installing source package ‘Rhtslib’ …
** using non-staged installation via StagedInstall field
** libs
cd “htslib-1.7” && make -f “/spack/apps/linux-centos7-x86_64/gcc-8.3.0/r-4.0.0-jfy3icn4kexk7kyabcoxuio2iyyww3o7/rlib/R/etc/Makeconf” -f “Makefile.Rhtslib”
make[1]: Entering directory /tmp/RtmpyhczcE/R.INSTALL51156f5ec932/Rhtslib/src/htslib-1.7' Makefile.Rhtslib:128: warning: overriding recipe for target .c.o’
/spack/apps/linux-centos7-x86_64/gcc-8.3.0/r-4.0.0-jfy3icn4kexk7kyabcoxuio2iyyww3o7/rlib/R/etc/Makeconf:167: warning: ignoring old recipe for target .c.o' /spack/apps/gcc/8.3.0/bin/gcc -g -O2 -fpic -I. -I/usr/local/include -D_FILE_OFFSET_BITS=64 -c -o kfunc.o kfunc.c /spack/apps/gcc/8.3.0/bin/gcc -g -O2 -fpic -I. -I/usr/local/include -D_FILE_OFFSET_BITS=64 -c -o knetfile.o knetfile.c /spack/apps/gcc/8.3.0/bin/gcc -g -O2 -fpic -I. -I/usr/local/include -D_FILE_OFFSET_BITS=64 -c -o kstring.o kstring.c /spack/apps/gcc/8.3.0/bin/gcc -g -O2 -fpic -I. -I/usr/local/include -D_FILE_OFFSET_BITS=64 -c -o bcf_sr_sort.o bcf_sr_sort.c /spack/apps/gcc/8.3.0/bin/gcc -g -O2 -fpic -I. -I/usr/local/include -D_FILE_OFFSET_BITS=64 -c -o bgzf.o bgzf.c In file included from bgzf.c:39: htslib/bgzf.h:35:10: fatal error: zlib.h: No such file or directory #include <zlib.h> ^~~~~~~~ compilation terminated. make[1]: *** [bgzf.o] Error 1 make[1]: Leaving directory /tmp/RtmpyhczcE/R.INSTALL51156f5ec932/Rhtslib/src/htslib-1.7’
make: *** [htslib] Error 2
ERROR: compilation failed for package ‘Rhtslib’
I’ve found that when R packages have prerequisite software you can modify ~/.R/Makevars and set important environment variables. For some reason setting them in your shell does not work.
I also took a look at Rhtslib and found that part of the installation process ignored settings in ~/.R/Makevars so I had to download the source code and modify Rhtslib/src/htslib-1.7/Makefile.Rhtslib
I downloaded the Rhtslib package into my scratch_R folder, which is the default folder that I set up for installing R packages.
Also, you mentioned adding a new line of code into ~/.R/Makevars. I did not have this .R folder in my home directory, so I created one manually.
Hi Cesar,
I was able to get minfi to work in a conda environment. Following this post https://bioconda.github.io/user/install.html
I create a new environment r_env: conda create --name r_env -c r r-essentials, then load bioconda, and finally conda install bioconductor-minfi. Still not sure why BiocManager::install(“minfi”) didn’t work for me, but glad that minfi is available in conda.
HI Cesar,
I am having similar issue as Keren faced. I followed the steps but I don’t see ~/.R/Makevars.
Please let me know how should I proceed. Another thing I have started installing required R package in my home directory but afraid that might be not sufficient space wise because I will be installing many bioconductor tools which needs lots of space. Do you think I should link my R in another project folder first or I can get more space for my home folder without any cost.
Thank you,
Priya
You’ll have to create ~/.R/Makevars if it doesn’t already exist.
In regards to where to save your R packages, you can run myquota to check how much space you have left. Home directory quotas are now 100GB so that should be plenty of space.
If you’re still concerned you can always symlink ~/R to one of your project directories like how we used to on the old cluster. If you want help with that process, please create a new topic.
Hi Cesar,
I have created .R in my home directory. Here are the steps just to make sure that I am doing it right.
mkdir .R
vim .R/Makevars
LDFLAGS=-L$(CURL_ROOT)/lib -L$(BZIP2_ROOT)/lib -L$(ZLIB_ROOT)/lib
and then
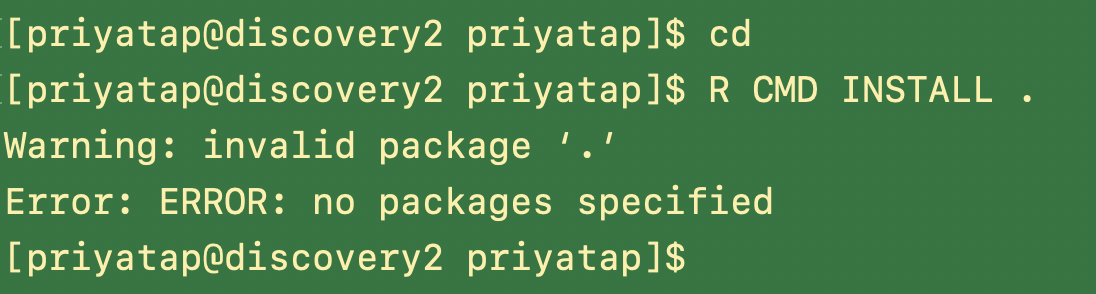
cd
R CMD INSTALL .
I got an error:
I’ve installed Rhtslib in the system library and it will sync over to Discovery in an hour or so. Once that’s available, you will then be able install other Bioconductor packages that depend on it like normal.
The Makevars file still appears to be ignored when compiling the package from source, or at least for installing other packages that depend on Rhtslib (which will error). To be explicit with the library paths, on line 45 of Rhtslib/src/htslib-1.7/Makefile.Rhtslib, beginning with LDFLAGS, uncomment and modify the line to be: