I am trying to build Smart-seq2 Multi-Sample pipeline which run on a WDL-compatible execution engine such Cromwell. I installed Cromwell with this tutorial and configured the backend providers for SLURM or Singularity+Slurm based on those examples. However each time I run the pipeline it failed because it cannot assign new job execution during the call-to-backend assignements. Did anyone succeed in running a pipeline on Cromwell? Would need suggestions for Cromwell installation and backend configuration. Thank you!
Hi,
I had some attempts to run GATK workflows via singularity container through Cromwell.
I think I got Cromwell to work.
I had to prepare local cache for singularity and download all containers beforehand. I did it via small shell script that I later sourced in Cromwell config.
/scratch2/ttrojan/set_singularity_cachedir.sh
#!/bin/bash
export SINGULARITY_CACHEDIR=/scratch2/ttrojan/singularity-cache
export SINGULARITY_TMPDIR=$SINGULARITY_CACHEDIR/tmp
export SINGULARITY_PULLDIR=$SINGULARITY_CACHEDIR/pull
export CWL_SINGULARITY_CACHE=$SINGULARITY_PULLDIR
I then set up Cromwell config in file /scratch2/ttrojan/cromwellslurmsingularity.conf
# This is a "default" Cromwell example that is intended for you you to start with
# and edit for your needs. Specifically, you will be interested to customize
# the configuration based on your preferred backend (see the backends section
# below in the file). For backend-specific examples for you to copy paste here,
# please see the cromwell.backend.examples folder in the repository. The files
# there also include links to online documentation (if it exists)
# This line is required. It pulls in default overrides from the embedded cromwell
# `reference.conf` (in core/src/main/resources) needed for proper performance of cromwell.
include required(classpath("application"))
# Cromwell HTTP server settings
webservice {
#port = 8000
#interface = 0.0.0.0
#binding-timeout = 5s
#instance.name = "reference"
}
# Cromwell "system" settings
system {
# If 'true', a SIGINT will trigger Cromwell to attempt to abort all currently running jobs before exiting
#abort-jobs-on-terminate = false
# If 'true', a SIGTERM or SIGINT will trigger Cromwell to attempt to gracefully shutdown in server mode,
# in particular clearing up all queued database writes before letting the JVM shut down.
# The shutdown is a multi-phase process, each phase having its own configurable timeout. See the Dev Wiki for more details.
#graceful-server-shutdown = true
# Cromwell will cap the number of running workflows at N
#max-concurrent-workflows = 5000
# Cromwell will launch up to N submitted workflows at a time, regardless of how many open workflow slots exist
#max-workflow-launch-count = 50
# Number of seconds between workflow launches
#new-workflow-poll-rate = 20
# Since the WorkflowLogCopyRouter is initialized in code, this is the number of workers
#number-of-workflow-log-copy-workers = 10
# Default number of cache read workers
#number-of-cache-read-workers = 25
io {
# throttle {
# # Global Throttling - This is mostly useful for GCS and can be adjusted to match
# # the quota availble on the GCS API
# #number-of-requests = 100000
# #per = 100 seconds
# }
# Number of times an I/O operation should be attempted before giving up and failing it.
#number-of-attempts = 5
}
# Maximum number of input file bytes allowed in order to read each type.
# If exceeded a FileSizeTooBig exception will be thrown.
input-read-limits {
#lines = 128000
#bool = 7
#int = 19
#float = 50
#string = 128000
#json = 128000
#tsv = 128000
#map = 128000
#object = 128000
}
abort {
# These are the default values in Cromwell, in most circumstances there should not be a need to change them.
# How frequently Cromwell should scan for aborts.
scan-frequency: 30 seconds
# The cache of in-progress aborts. Cromwell will add entries to this cache once a WorkflowActor has been messaged to abort.
# If on the next scan an 'Aborting' status is found for a workflow that has an entry in this cache, Cromwell will not ask
# the associated WorkflowActor to abort again.
cache {
enabled: true
# Guava cache concurrency.
concurrency: 1
# How long entries in the cache should live from the time they are added to the cache.
ttl: 20 minutes
# Maximum number of entries in the cache.
size: 100000
}
}
# Cromwell reads this value into the JVM's `networkaddress.cache.ttl` setting to control DNS cache expiration
dns-cache-ttl: 3 minutes
}
docker {
hash-lookup {
# Set this to match your available quota against the Google Container Engine API
#gcr-api-queries-per-100-seconds = 1000
# Time in minutes before an entry expires from the docker hashes cache and needs to be fetched again
#cache-entry-ttl = "20 minutes"
# Maximum number of elements to be kept in the cache. If the limit is reached, old elements will be removed from the cache
#cache-size = 200
# How should docker hashes be looked up. Possible values are "local" and "remote"
# "local": Lookup hashes on the local docker daemon using the cli
# "remote": Lookup hashes on docker hub, gcr, gar, quay
#method = "remote"
enabled = "false"
}
}
# Here is where you can define the backend providers that Cromwell understands.
# The default is a local provider.
# To add additional backend providers, you should copy paste additional backends
# of interest that you can find in the cromwell.example.backends folder
# folder at https://www.github.com/broadinstitute/cromwell
# Other backend providers include SGE, SLURM, Docker, udocker, Singularity. etc.
# Don't forget you will need to customize them for your particular use case.
backend {
# Override the default backend.
default = slurm
# The list of providers.
providers {
# Copy paste the contents of a backend provider in this section
# Examples in cromwell.example.backends include:
# LocalExample: What you should use if you want to define a new backend provider
# AWS: Amazon Web Services
# BCS: Alibaba Cloud Batch Compute
# TES: protocol defined by GA4GH
# TESK: the same, with kubernetes support
# Google Pipelines, v2 (PAPIv2)
# Docker
# Singularity: a container safe for HPC
# Singularity+Slurm: and an example on Slurm
# udocker: another rootless container solution
# udocker+slurm: also exemplified on slurm
# HtCondor: workload manager at UW-Madison
# LSF: the Platform Load Sharing Facility backend
# SGE: Sun Grid Engine
# SLURM: workload manager
# Note that these other backend examples will need tweaking and configuration.
# Please open an issue https://www.github.com/broadinstitute/cromwell if you have any questions
slurm {
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory"
config {
# Root directory where Cromwell writes job results in the container. This value
# can be used to specify where the execution folder is mounted in the container.
# it is used for the construction of the docker_cwd string in the submit-docker
# value above.
dockerRoot = "/cromwell-executions"
concurrent-job-limit = 10
# If an 'exit-code-timeout-seconds' value is specified:
# - check-alive will be run at this interval for every job
# - if a job is found to be not alive, and no RC file appears after this interval
# - Then it will be marked as Failed.
## Warning: If set, Cromwell will run 'check-alive' for every job at this interval
exit-code-timeout-seconds = 360
filesystems {
local {
localization: [
# soft link does not work for docker with --contain. Hard links won't work
# across file systems
"copy", "hard-link", "soft-link"
]
caching {
duplication-strategy: ["copy", "hard-link", "soft-link"]
hashing-strategy: "file"
}
}
}
#
runtime-attributes = """
Int runtime_minutes = 600
Int cpus = 2
Int requested_memory_mb_per_core = 8000
String? docker
String? partition
String? account
"""
submit = """
sbatch \
--wait \
-J ${job_name} \
-D ${cwd} \
-o ${out} \
-e ${err} \
-t ${runtime_minutes} \
${"-c " + cpus} \
--mem-per-cpu=${requested_memory_mb_per_core} \
--partition=${partition} \
--account=${account} \
--wrap "/bin/bash ${script}"
"""
submit-docker = """
# SINGULARITY_CACHEDIR needs to point to a directory accessible by
# the jobs (i.e. not lscratch). Might want to use a workflow local
# cache dir like in run.sh
source /scratch2/ttrojan/set_singularity_cachedir.sh
SINGULARITY_CACHEDIR=/scratch2/ttrojan/singularity-cache
echo "SINGULARITY_CACHEDIR $SINGULARITY_CACHEDIR"
if [ -z $SINGULARITY_CACHEDIR ]; then
CACHE_DIR=$HOME/.singularity
else
CACHE_DIR=$SINGULARITY_CACHEDIR
fi
mkdir -p $CACHE_DIR
echo "SINGULARITY_CACHEDIR $SINGULARITY_CACHEDIR"
LOCK_FILE=$CACHE_DIR/singularity_pull_flock
# we want to avoid all the cromwell tasks hammering each other trying
# to pull the container into the cache for the first time. flock works
# on GPFS, netapp, and vast (of course only for processes on the same
# machine which is the case here since we're pulling it in the master
# process before submitting).
#flock --exclusive --timeout 1200 $LOCK_FILE \
# singularity exec --containall docker://${docker} \
# echo "successfully pulled ${docker}!" &> /dev/null
# Ensure singularity is loaded if it's installed as a module
#module load Singularity/3.0.1
# Build the Docker image into a singularity image
IMAGE=${docker}.sif
singularity build $IMAGE docker://${docker}
# Submit the script to SLURM
sbatch \
--wait \
-J ${job_name} \
-D ${cwd} \
-o ${cwd}/execution/stdout \
-e ${cwd}/execution/stderr \
-t ${runtime_minutes} \
${"-c " + cpus} \
--mem-per-cpu=${requested_memory_mb_per_core} \
--partition=${partition} \
--account=${account} \
--wrap "singularity exec --bind ${cwd}:${docker_cwd} ${docker}.sif ${job_shell} ${docker_script}"
"""
kill = "scancel ${job_id}"
check-alive = "squeue -j ${job_id}"
job-id-regex = "Submitted batch job (\\d+).*"
}
}
}
}
The following was my submission script:
#!/bin/bash
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=12
#SBATCH --mem-per-cpu=6g
#SBATCH --time=16:00:00
#SBATCH --partition=epyc-64
#SBATCH --account=ttrojan_123
module purge
module load usc
module load openjdk
java -jar -Dconfig.file=cromwell.conf cromwell-71.jar run gatktest.wdl -i gatktest_inputs.json
gatktest.wdl:
workflow helloCountBasesCaller {
call CountBasesCaller
}
task CountBasesCaller {
String GATKcontainer
String sampleName
String partition
String account
File inputBAM
command {
gatk \
CountBases \
-I ${inputBAM} \
> ${sampleName}.txt
}
output {
File rawTXT = "${sampleName}.txt"
}
runtime {
docker: "${GATKcontainer}"
partition: "${partition}"
account: "${account}"
}
}
and finally gatktest_inputs.json
{
"helloCountBasesCaller.CountBasesCaller.inputBAM": "NA12878.bam",
"helloCountBasesCaller.CountBasesCaller.sampleName": "outdata",
"helloCountBasesCaller.CountBasesCaller.GATKcontainer": "broadinstitute/gatk:4.2.3.0",
"helloCountBasesCaller.CountBasesCaller.partition": "epyc-64",
"helloCountBasesCaller.CountBasesCaller.account": "ttrojan_123"
}
I edited paths and account name for this post.
Thanks for submitting this issue, as putting some Cromwell guide is on my todo list for this year
Please let me know if the above works for you
1 Like
Dear Osinski,
Thank you very much for your help! I tried your suggestions (by updating path and account name) and unfortunately it didn’t work! Here is what the output look like (I edited shorten as it was very long):
[2022-04-12 09:13:14,65] [info] MaterializeWorkflowDescriptorActor [8732d135]: Call-to-Backend assignments: SmartSeq2SingleCell.CollectMultipleMetrics -> slurm, SmartSeq2SingleCell.HISAT2SingleEndTranscriptome -> slurm, SmartSeq2SingleCell.HISAT2Transcriptome -> slurm, SmartSeq2SingleCell.GroupQCOutputs -> slurm, MultiSampleSmartSeq2.checkArrays -> slurm, SmartSeq2SingleCell.SmartSeq2LoomOutput -> slurm, SmartSeq2SingleCell.CollectRnaMetrics -> slurm, SmartSeq2SingleCell.HISAT2PairedEnd -> slurm, SmartSeq2SingleCell.CollectDuplicationMetrics -> slurm, SmartSeq2SingleCell.HISAT2SingleEnd -> slurm, MultiSampleSmartSeq2.AggregateLoom -> slurm, SmartSeq2SingleCell.RSEMExpression -> slurm
[2022-04-12 09:13:14,96] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [cpu, memory, disks] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:14,97] [warn] slurm [8732d135]: Key/s [preemptible, disks, cpu, memory] is/are not supported by backend. Unsupported attributes will not be part of job executions.
[2022-04-12 09:13:17,02] [info] Not triggering log of restart checking token queue status. Effective log interval = None
[2022-04-12 09:13:17,08] [info] Not triggering log of execution token queue status. Effective log interval = None
[2022-04-12 09:13:17,21] [info] WorkflowExecutionActor-8732d135-ce1e-4605-abe1-58f929664c44 [8732d135]: Starting MultiSampleSmartSeq2.checkArrays
[2022-04-12 09:13:22,10] [info] Assigned new job execution tokens to the following groups: 8732d135: 1
[2022-04-12 09:13:22,24] [warn] DispatchedConfigAsyncJobExecutionActor [8732d135MultiSampleSmartSeq2.checkArrays:NA:1]: Unrecognized runtime attribute keys: disks, cpu, memory
[2022-04-12 09:13:22,31] [info] DispatchedConfigAsyncJobExecutionActor [8732d135MultiSampleSmartSeq2.checkArrays:NA:1]: set -e
[2022-04-12 09:13:22,37] [info] DispatchedConfigAsyncJobExecutionActor [8732d135MultiSampleSmartSeq2.checkArrays:NA:1]: executing: # SINGULARITY_CACHEDIR needs to point to a directory accessible by
# the jobs (i.e. not lscratch). Might want to use a workflow local
# cache dir like in run.sh
source /scratch1/trojan/cromwell/set_singularity_cachedir.sh
SINGULARITY_CACHEDIR=/scratch1/trojan/cromwell/singularity-cache
echo "SINGULARITY_CACHEDIR $SINGULARITY_CACHEDIR"
[2022-04-12 09:13:45,88] [info] DispatchedConfigAsyncJobExecutionActor [c7431018SmartSeq2SingleCell.HISAT2Transcriptome:NA:1]: executing: # SINGULARITY_CACHEDIR needs to point to a directory accessible by
# the jobs (i.e. not lscratch). Might want to use a workflow local
# cache dir like in run.sh
source /scratch1/trojan/cromwell/set_singularity_cachedir.sh
SINGULARITY_CACHEDIR=/scratch1/trojan/cromwell/singularity-cache
echo "SINGULARITY_CACHEDIR $SINGULARITY_CACHEDIR"
if [ -z $SINGULARITY_CACHEDIR ]; then
CACHE_DIR=$HOME/.singularity
else
CACHE_DIR=$SINGULARITY_CACHEDIR
fi
mkdir -p $CACHE_DIR
echo "SINGULARITY_CACHEDIR $SINGULARITY_CACHEDIR"
LOCK_FILE=$CACHE_DIR/singularity_pull_flock
# we want to avoid all the cromwell tasks hammering each other trying
# to pull the container into the cache for the first time. flock works
# on GPFS, netapp, and vast (of course only for processes on the same
# machine which is the case here since we're pulling it in the master
# process before submitting).
#flock --exclusive --timeout 1200 $LOCK_FILE \
# singularity exec --containall docker://quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0 \
# echo "successfully pulled quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0!" &> /dev/null
# Ensure singularity is loaded if it's installed as a module
#module load Singularity/3.0.1
# Build the Docker image into a singularity image
IMAGE=quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0.sif
singularity build $IMAGE docker://quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0
# Submit the script to SLURM
sbatch \
--wait \
-J cromwell_c7431018_HISAT2Transcriptome \
-D /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-1/SmartSeq2SingleCell/c7431018-82c3-4804-9c97-a465036396d5/call-HISAT2Transcriptome \
-o /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-1/SmartSeq2SingleCell/c7431018-82c3-4804-9c97-a465036396d5/call-HISAT2Transcriptome/execution/stdout \
-e /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-1/SmartSeq2SingleCell/c7431018-82c3-4804-9c97-a465036396d5/call-HISAT2Transcriptome/execution/stderr \
-t 600 \
-c 2 \
--mem-per-cpu=8000 \
--partition= \
--account= \
--wrap "singularity exec --bind /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-1/SmartSeq2SingleCell/c7431018-82c3-4804-9c97-a465036396d5/call-HISAT2Transcriptome:/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-1/SmartSeq2SingleCell/c7431018-82c3-4804-9c97-a465036396d5/call-HISAT2Transcriptome quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0.sif /bin/bash /cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-1/SmartSeq2SingleCell/c7431018-82c3-4804-9c97-a465036396d5/call-HISAT2Transcriptome/execution/script"
[2022-04-12 09:13:52,67] [info] DispatchedConfigAsyncJobExecutionActor [857ab287SmartSeq2SingleCell.HISAT2Transcriptome:NA:1]: set -e
[2022-04-12 09:14:22,61] [info] WorkflowManagerActor: Workflow 8732d135-ce1e-4605-abe1-58f929664c44 failed (during ExecutingWorkflowState): java.lang.RuntimeException: Unable to start job. Check the stderr file for possible errors: /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-checkArrays/execution/stderr.submit
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.$anonfun$execute$2(SharedFileSystemAsyncJobExecutionActor.scala:165)
at scala.util.Either.fold(Either.scala:191)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute(SharedFileSystemAsyncJobExecutionActor.scala:160)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute$(SharedFileSystemAsyncJobExecutionActor.scala:155)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.execute(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.$anonfun$executeAsync$1(StandardAsyncExecutionActor.scala:748)
at scala.util.Try$.apply(Try.scala:213)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync$(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeAsync(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover(StandardAsyncExecutionActor.scala:1138)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover$(StandardAsyncExecutionActor.scala:1130)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeOrRecover(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.async.AsyncBackendJobExecutionActor.$anonfun$robustExecuteOrRecover$1(AsyncBackendJobExecutionActor.scala:65)
at cromwell.core.retry.Retry$.withRetry(Retry.scala:46)
at cromwell.backend.async.AsyncBackendJobExecutionActor.withRetry(AsyncBackendJobExecutionActor.scala:61)
at cromwell.backend.async.AsyncBackendJobExecutionActor.cromwell$backend$async$AsyncBackendJobExecutionActor$$robustExecuteOrRecover(AsyncBackendJobExecutionActor.scala:65)
at cromwell.backend.async.AsyncBackendJobExecutionActor$$anonfun$receive$1.applyOrElse(AsyncBackendJobExecutionActor.scala:88)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at akka.actor.Actor.aroundReceive(Actor.scala:539)
at akka.actor.Actor.aroundReceive$(Actor.scala:537)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.aroundReceive(ConfigAsyncJobExecutionActor.scala:215)
at akka.actor.ActorCell.receiveMessage(ActorCell.scala:614)
at akka.actor.ActorCell.invoke(ActorCell.scala:583)
at akka.dispatch.Mailbox.processMailbox(Mailbox.scala:268)
at akka.dispatch.Mailbox.run(Mailbox.scala:229)
at akka.dispatch.Mailbox.exec(Mailbox.scala:241)
at akka.dispatch.forkjoin.ForkJoinTask.doExec(ForkJoinTask.java:260)
at akka.dispatch.forkjoin.ForkJoinPool$WorkQueue.runTask(ForkJoinPool.java:1339)
at akka.dispatch.forkjoin.ForkJoinPool.runWorker(ForkJoinPool.java:1979)
at akka.dispatch.forkjoin.ForkJoinWorkerThread.run(ForkJoinWorkerThread.java:107)
java.lang.RuntimeException: Unable to start job. Check the stderr file for possible errors: /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-1/SmartSeq2SingleCell/c7431018-82c3-4804-9c97-a465036396d5/call-HISAT2Transcriptome/execution/stderr.submit
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.$anonfun$execute$2(SharedFileSystemAsyncJobExecutionActor.scala:165)
at scala.util.Either.fold(Either.scala:191)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute(SharedFileSystemAsyncJobExecutionActor.scala:160)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute$(SharedFileSystemAsyncJobExecutionActor.scala:155)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.execute(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.$anonfun$executeAsync$1(StandardAsyncExecutionActor.scala:748)
at scala.util.Try$.apply(Try.scala:213)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync$(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeAsync(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover(StandardAsyncExecutionActor.scala:1138)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover$(StandardAsyncExecutionActor.scala:1130)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeOrRecover(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.async.AsyncBackendJobExecutionActor.$anonfun$robustExecuteOrRecover$1(AsyncBackendJobExecutionActor.scala:65)
at cromwell.core.retry.Retry$.withRetry(Retry.scala:46)
at cromwell.backend.async.AsyncBackendJobExecutionActor.withRetry(AsyncBackendJobExecutionActor.scala:61)
at cromwell.backend.async.AsyncBackendJobExecutionActor.cromwell$backend$async$AsyncBackendJobExecutionActor$$robustExecuteOrRecover(AsyncBackendJobExecutionActor.scala:65)
at cromwell.backend.async.AsyncBackendJobExecutionActor$$anonfun$receive$1.applyOrElse(AsyncBackendJobExecutionActor.scala:88)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at akka.actor.Actor.aroundReceive(Actor.scala:539)
at akka.actor.Actor.aroundReceive$(Actor.scala:537)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.aroundReceive(ConfigAsyncJobExecutionActor.scala:215)
at akka.actor.ActorCell.receiveMessage(ActorCell.scala:614)
at akka.actor.ActorCell.invoke(ActorCell.scala:583)
at akka.dispatch.Mailbox.processMailbox(Mailbox.scala:268)
at akka.dispatch.Mailbox.run(Mailbox.scala:229)
at akka.dispatch.Mailbox.exec(Mailbox.scala:241)
at akka.dispatch.forkjoin.ForkJoinTask.doExec(ForkJoinTask.java:260)
at akka.dispatch.forkjoin.ForkJoinPool$WorkQueue.runTask(ForkJoinPool.java:1339)
at akka.dispatch.forkjoin.ForkJoinPool.runWorker(ForkJoinPool.java:1979)
at akka.dispatch.forkjoin.ForkJoinWorkerThread.run(ForkJoinWorkerThread.java:107)
java.lang.RuntimeException: Unable to start job. Check the stderr file for possible errors: /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-0/SmartSeq2SingleCell/857ab287-ac3e-4978-bf43-767bc78c640a/call-HISAT2Transcriptome/execution/stderr.submit
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.$anonfun$execute$2(SharedFileSystemAsyncJobExecutionActor.scala:165)
at scala.util.Either.fold(Either.scala:191)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute(SharedFileSystemAsyncJobExecutionActor.scala:160)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute$(SharedFileSystemAsyncJobExecutionActor.scala:155)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.execute(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.$anonfun$executeAsync$1(StandardAsyncExecutionActor.scala:748)
at scala.util.Try$.apply(Try.scala:213)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync$(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeAsync(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover(StandardAsyncExecutionActor.scala:1138)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover$(StandardAsyncExecutionActor.scala:1130)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeOrRecover(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.async.AsyncBackendJobExecutionActor.$anonfun$robustExecuteOrRecover$1(AsyncBackendJobExecutionActor.scala:65)
at cromwell.core.retry.Retry$.withRetry(Retry.scala:46)
at cromwell.backend.async.AsyncBackendJobExecutionActor.withRetry(AsyncBackendJobExecutionActor.scala:61)
at cromwell.backend.async.AsyncBackendJobExecutionActor.cromwell$backend$async$AsyncBackendJobExecutionActor$$robustExecuteOrRecover(AsyncBackendJobExecutionActor.scala:65)
at cromwell.backend.async.AsyncBackendJobExecutionActor$$anonfun$receive$1.applyOrElse(AsyncBackendJobExecutionActor.scala:88)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at akka.actor.Actor.aroundReceive(Actor.scala:539)
at akka.actor.Actor.aroundReceive$(Actor.scala:537)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.aroundReceive(ConfigAsyncJobExecutionActor.scala:215)
at akka.actor.ActorCell.receiveMessage(ActorCell.scala:614)
at akka.actor.ActorCell.invoke(ActorCell.scala:583)
at akka.dispatch.Mailbox.processMailbox(Mailbox.scala:268)
at akka.dispatch.Mailbox.run(Mailbox.scala:229)
at akka.dispatch.Mailbox.exec(Mailbox.scala:241)
at akka.dispatch.forkjoin.ForkJoinTask.doExec(ForkJoinTask.java:260)
at akka.dispatch.forkjoin.ForkJoinPool$WorkQueue.runTask(ForkJoinPool.java:1339)
at akka.dispatch.forkjoin.ForkJoinPool.runWorker(ForkJoinPool.java:1979)
at akka.dispatch.forkjoin.ForkJoinWorkerThread.run(ForkJoinWorkerThread.java:107)
java.lang.RuntimeException: Unable to start job. Check the stderr file for possible errors: /scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/8732d135-ce1e-4605-abe1-58f929664c44/call-sc_pe/shard-0/SmartSeq2SingleCell/857ab287-ac3e-4978-bf43-767bc78c640a/call-HISAT2PairedEnd/execution/stderr.submit
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.$anonfun$execute$2(SharedFileSystemAsyncJobExecutionActor.scala:165)
at scala.util.Either.fold(Either.scala:191)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute(SharedFileSystemAsyncJobExecutionActor.scala:160)
at cromwell.backend.sfs.SharedFileSystemAsyncJobExecutionActor.execute$(SharedFileSystemAsyncJobExecutionActor.scala:155)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.execute(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.$anonfun$executeAsync$1(StandardAsyncExecutionActor.scala:748)
at scala.util.Try$.apply(Try.scala:213)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeAsync$(StandardAsyncExecutionActor.scala:748)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeAsync(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover(StandardAsyncExecutionActor.scala:1138)
at cromwell.backend.standard.StandardAsyncExecutionActor.executeOrRecover$(StandardAsyncExecutionActor.scala:1130)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.executeOrRecover(ConfigAsyncJobExecutionActor.scala:215)
at cromwell.backend.async.AsyncBackendJobExecutionActor.$anonfun$robustExecuteOrRecover$1(AsyncBackendJobExecutionActor.scala:65)
at cromwell.core.retry.Retry$.withRetry(Retry.scala:46)
at cromwell.backend.async.AsyncBackendJobExecutionActor.withRetry(AsyncBackendJobExecutionActor.scala:61)
at cromwell.backend.async.AsyncBackendJobExecutionActor.cromwell$backend$async$AsyncBackendJobExecutionActor$$robustExecuteOrRecover(AsyncBackendJobExecutionActor.scala:65)
at cromwell.backend.async.AsyncBackendJobExecutionActor$$anonfun$receive$1.applyOrElse(AsyncBackendJobExecutionActor.scala:88)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at scala.PartialFunction$OrElse.applyOrElse(PartialFunction.scala:176)
at akka.actor.Actor.aroundReceive(Actor.scala:539)
at akka.actor.Actor.aroundReceive$(Actor.scala:537)
at cromwell.backend.impl.sfs.config.DispatchedConfigAsyncJobExecutionActor.aroundReceive(ConfigAsyncJobExecutionActor.scala:215)
at akka.actor.ActorCell.receiveMessage(ActorCell.scala:614)
at akka.actor.ActorCell.invoke(ActorCell.scala:583)
at akka.dispatch.Mailbox.processMailbox(Mailbox.scala:268)
at akka.dispatch.Mailbox.run(Mailbox.scala:229)
at akka.dispatch.Mailbox.exec(Mailbox.scala:241)
at akka.dispatch.forkjoin.ForkJoinTask.doExec(ForkJoinTask.java:260)
at akka.dispatch.forkjoin.ForkJoinPool$WorkQueue.runTask(ForkJoinPool.java:1339)
at akka.dispatch.forkjoin.ForkJoinPool.runWorker(ForkJoinPool.java:1979)
at akka.dispatch.forkjoin.ForkJoinWorkerThread.run(ForkJoinWorkerThread.java:107)
To my understanding it failed to start jobs. This is my sh script:
#!/bin/bash
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem=32GB
#SBATCH --time=2:00:00
#SBATCH --mail-type=ALL
#SBATCH --account=trojan_123
#SBATCH --mail-user=trojan@usc.edu
module purge
module load usc
module load openjdk
java -Dconfig.file=cromwellslurmsingularity.conf -jar cromwell-78.jar run MultiSampleSmartSeq2_v2.2.9.wdl -i MultiSampleSmartSeq2_v2.2.9.options.json
MultiSampleSmartSeq2_v2.2.9.wdl
version 1.0
import "SmartSeq2SingleSample.wdl" as single_cell_run
import "LoomUtils.wdl" as LoomUtils
workflow MultiSampleSmartSeq2 {
meta {
description: "The MultiSampleSmartSeq2 pipeline runs multiple SS2 samples in a single pipeline invocation"
allowNestedInputs: true
}
input {
# Gene Annotation
File genome_ref_fasta
File rrna_intervals
File gene_ref_flat
# Reference index information
File hisat2_ref_name
File hisat2_ref_trans_name
File hisat2_ref_index
File hisat2_ref_trans_index
File rsem_ref_index
# Sample information
String stranded
Array[String] input_ids
Array[String]? input_names
Array[String] fastq1_input_files
Array[String] fastq2_input_files = []
String batch_id
String? batch_name
Array[String]? project_id
Array[String]? project_name
Array[String]? library
Array[String]? species
Array[String]? organ
String? input_name_metadata_field
String? input_id_metadata_field
Boolean paired_end
}
# Version of this pipeline
String pipeline_version = "2.2.9"
if (false) {
String? none = "None"
}
# Parameter metadata information
parameter_meta {
genome_ref_fasta: "Genome reference in fasta format"
rrna_intervals: "rRNA interval file required by Picard"
gene_ref_flat: "Gene refflat file required by Picard"
hisat2_ref_name: "HISAT2 reference index name"
hisat2_ref_trans_name: "HISAT2 transcriptome index file name"
hisat2_ref_index: "HISAT2 reference index file in tarball"
hisat2_ref_trans_index: "HISAT2 transcriptome index file in tarball"
rsem_ref_index: "RSEM reference index file in tarball"
stranded: "Library strand information example values: FR RF NONE"
input_ids: "Array of input ids"
input_names: "Array of input names"
input_id_metadata_field: "String that describes the metadata field containing the input_ids"
input_name_metadata_field: "String that describes the metadata field containing the input_names"
fastq1_input_files: "Array of fastq1 files; order must match the order in input_id."
fastq2_input_files: "Array of fastq2 files for paired end runs; order must match fastq1_input_files and input_id."
batch_id: " Identifier for the batch"
paired_end: "Is the sample paired end or not"
}
# Check that all input arrays are the same length
call checkInputArrays as checkArrays{
input:
paired_end = paired_end,
input_ids = input_ids,
input_names = input_names,
fastq1_input_files = fastq1_input_files,
fastq2_input_files = fastq2_input_files
}
### Execution starts here ###
if (paired_end) {
scatter(idx in range(length(input_ids))) {
call single_cell_run.SmartSeq2SingleCell as sc_pe {
input:
fastq1 = fastq1_input_files[idx],
fastq2 = fastq2_input_files[idx],
stranded = stranded,
genome_ref_fasta = genome_ref_fasta,
rrna_intervals = rrna_intervals,
gene_ref_flat = gene_ref_flat,
hisat2_ref_index = hisat2_ref_index,
hisat2_ref_name = hisat2_ref_name,
hisat2_ref_trans_index = hisat2_ref_trans_index,
hisat2_ref_trans_name = hisat2_ref_trans_name,
rsem_ref_index = rsem_ref_index,
input_id = input_ids[idx],
output_name = input_ids[idx],
paired_end = paired_end,
input_name_metadata_field = input_name_metadata_field,
input_id_metadata_field = input_id_metadata_field,
input_name = if defined(input_names) then select_first([input_names])[idx] else none
}
}
}
if (!paired_end) {
scatter(idx in range(length(input_ids))) {
call single_cell_run.SmartSeq2SingleCell as sc_se {
input:
fastq1 = fastq1_input_files[idx],
stranded = stranded,
genome_ref_fasta = genome_ref_fasta,
rrna_intervals = rrna_intervals,
gene_ref_flat = gene_ref_flat,
hisat2_ref_index = hisat2_ref_index,
hisat2_ref_name = hisat2_ref_name,
hisat2_ref_trans_index = hisat2_ref_trans_index,
hisat2_ref_trans_name = hisat2_ref_trans_name,
rsem_ref_index = rsem_ref_index,
input_id = input_ids[idx],
output_name = input_ids[idx],
paired_end = paired_end,
input_name_metadata_field = input_name_metadata_field,
input_id_metadata_field = input_id_metadata_field,
input_name = if defined(input_names) then select_first([input_names])[idx] else none
}
}
}
Array[File] loom_output_files = select_first([sc_pe.loom_output_files, sc_se.loom_output_files])
Array[File] bam_files_intermediate = select_first([sc_pe.aligned_bam, sc_se.aligned_bam])
Array[File] bam_index_files_intermediate = select_first([sc_pe.bam_index, sc_se.bam_index])
### Aggregate the Loom Files Directly ###
call LoomUtils.AggregateSmartSeq2Loom as AggregateLoom {
input:
loom_input = loom_output_files,
batch_id = batch_id,
batch_name = batch_name,
project_id = if defined(project_id) then select_first([project_id])[0] else none,
project_name = if defined(project_name) then select_first([project_name])[0] else none,
library = if defined(library) then select_first([library])[0] else none,
species = if defined(species) then select_first([species])[0] else none,
organ = if defined(organ) then select_first([organ])[0] else none,
pipeline_version = "MultiSampleSmartSeq2_v~{pipeline_version}"
}
### Pipeline output ###
output {
# Bam files and their indexes
Array[File] bam_files = bam_files_intermediate
Array[File] bam_index_files = bam_index_files_intermediate
File loom_output = AggregateLoom.loom_output_file
}
}
task checkInputArrays {
input {
Boolean paired_end
Array[String] input_ids
Array[String]? input_names
Array[String] fastq1_input_files
Array[String] fastq2_input_files
}
Int len_input_ids = length(input_ids)
Int len_fastq1_input_files = length(fastq1_input_files)
Int len_fastq2_input_files = length(fastq2_input_files)
Int len_input_names = if defined(input_names) then length(select_first([input_names])) else 0
meta {
description: "checks input arrays to ensure that all arrays are the same length"
}
command {
set -e
if [[ ~{len_input_ids} != ~{len_fastq1_input_files} ]]
then
echo "ERROR: Different number of arguments for input_id and fastq1 files"
exit 1;
fi
if [[ ~{len_input_names} != 0 && ~{len_input_ids} != ~{len_input_names} ]]
then
echo "ERROR: Different number of arguments for input_name and input_id"
exit 1;
fi
if ~{paired_end} && [[ ~{len_fastq2_input_files} != ~{len_input_ids} ]]
then
echo "ERROR: Different number of arguments for sample names and fastq1 files"
exit 1;
fi
exit 0;
}
runtime {
docker: "ubuntu:18.04"
cpu: 1
memory: "1 GiB"
disks: "local-disk 1 HDD"
}
}
and finally MultiSampleSmartSeq2_v2.2.9.options.json
{
"MultiSampleSmartSeq2.genome_ref_fasta": "GRCh38.primary_assembly.genome.fa",
"MultiSampleSmartSeq2.rrna_intervals": "gencode.v27.primary_assembly.annotation.interval_list",
"MultiSampleSmartSeq2.gene_ref_flat": "gencode.v27.primary_assembly.annotation.refflat.txt",
"MultiSampleSmartSeq2.hisat2_ref_name": "hisat2_primary_gencode_human_v27",
"MultiSampleSmartSeq2.hisat2_ref_trans_name": "hisat2_from_rsem_star_primary_gencode_human_v27",
"MultiSampleSmartSeq2.hisat2_ref_index": "hisat2_primary_gencode_human_v27.tar.gz",
"MultiSampleSmartSeq2.hisat2_ref_trans_index": "hisat2_from_rsem_star_primary_gencode_human_v27.tar.gz",
"MultiSampleSmartSeq2.rsem_ref_index": "rsem_primary_gencode_human_v27.tar",
"MultiSampleSmartSeq2.stranded": "NONE",
"MultiSampleSmartSeq2.paired_end": true,
"MultiSampleSmartSeq2.input_ids": ["B1", "B100"],
"MultiSampleSmartSeq2.fastq1_input_files": [
"B1_CKDL220004838-1a-AK31169-AK31170_HG5NCDSX3_L1_1.fq.gz",
"B100_CKDL220004838-1a-AK31158-AK31159_HG5NCDSX3_L1_1.fq.gz"
],
"MultiSampleSmartSeq2.fastq2_input_files": [
"B1_CKDL220004838-1a-AK31169-AK31170_HG5NCDSX3_L1_2.fq.gz",
"B100_CKDL220004838-1a-AK31158-AK31159_HG5NCDSX3_L1_2.fq.gz"
],
"MultiSampleSmartSeq2.batch_id": "ctc_paired_SSmulti"
}
Thank you for your help!
Hi,
I looked through your logs and I think you might be very close to get it to work.
The reason I had to set singularity to use cache is that compute nodes do not have access to the internet, so any container image has to be downloaded beforehand (using transfer nodes or login nodes)
We host office hours every Tuesday from 2.30 until 5. Maybe you can join today and we can go through troubleshooting Cromwell? https://www.carc.usc.edu/news-and-events/events
If not we can schedule individual consultation at another time
Thank you! I will try to join today!
Hello Osinski,
I thought I was about to get it work yesterday when I realized what you meant by “downloading the container image”. Indeed, I reviewed all the .wdl files used in the workflow and saw multiple argument like this one String docker = "quay.io/humancellatlas/secondary-analysis-rsem:v0.2.2-1.3.0"
So I downloaded all of them with singularity pull docker:// . I wasn’t sure where to store those containers and how to edit the .wdl files. Should I store then in singularity-cache folder?
I want to share one example of .wdl file (the workflow includes multiple tasks similar to this)
RSEM.wdl
version 1.0
task RSEMExpression {
input {
File trans_aligned_bam
File rsem_genome
String output_basename
Boolean is_paired
# runtime values
String docker = "quay.io/humancellatlas/secondary-analysis-rsem:v0.2.2-1.3.0"
Int machine_mem_mb = 32768
Int cpu = 4
# use provided disk number or dynamically size on our own, with 200GiB of additional disk
Int disk = ceil(size(trans_aligned_bam, "GiB") + size(rsem_genome, "GiB") + 200)
Int preemptible = 5
}
meta {
description: "This task will quantify gene expression matrix by using RSEM. The output include gene-level and isoform-level results."
}
parameter_meta {
trans_aligned_bam: "input transcriptome aligned bam"
rsem_genome: "tar'd RSEM genome"
output_basename: "basename used for output files"
docker: "(optional) the docker image containing the runtime environment for this task"
machine_mem_mb: "(optional) the amount of memory (MiB) to provision for this task"
cpu: "(optional) the number of cpus to provision for this task"
disk: "(optional) the amount of disk space (GiB) to provision for this task"
preemptible: "(optional) if non-zero, request a pre-emptible instance and allow for this number of preemptions before running the task on a non preemptible machine"
}
command {
set -e
tar --no-same-owner -xvf ${rsem_genome}
rsem-calculate-expression \
--bam \
${true="--paired-end" false="" is_paired} \
-p ${cpu} \
--time --seed 555 \
--calc-pme \
--single-cell-prior \
${trans_aligned_bam} \
rsem/rsem_trans_index \
"${output_basename}"
}
runtime {
docker: docker
memory: "${machine_mem_mb} MiB"
disks: "local-disk ${disk} HDD"
cpu: cpu
preemptible: preemptible
}
output {
File rsem_gene = "${output_basename}.genes.results"
File rsem_isoform = "${output_basename}.isoforms.results"
File rsem_time = "${output_basename}.time"
File rsem_cnt = "${output_basename}.stat/${output_basename}.cnt"
File rsem_model = "${output_basename}.stat/${output_basename}.model"
File rsem_theta = "${output_basename}.stat/${output_basename}.theta"
}
}
How should I replace the argument String docker = "quay.io/humancellatlas/secondary-analysis-rsem:v0.2.2-1.3.0" , docker: "(optional) the docker image containing the runtime environment for this task" and runtime { docker: docker ? In the input .json file there are no input for dockers paths.
Thank you very much for your help!
Setting up a singularity cache before downloading the container will place the container in that location.
When you use it in your workflow (of course, after sourcing the singularity cache) with singularity run singularity will try first checking the cache for the requested container version.
In other words:
On the login node:
- setup singularity cache
singularity pull docker://quay.io/humancellatlas/secondary-analysis-rsem:v0.2.2-1.3.0
On the computer node during the workflow execution:
- setup singularity cache
- standard
singularity run docker://quay.io/humancellatlas/secondary-analysis-rsem:v0.2.2-1.3.0should first try using the container from the cache
If you set the singularity cache and pulled the container it should be just picked up during the execution. If not, we can try modifying the Cromwell configuration file and use singularity images directly (harder to maintain with several workflows)
Cromwell and WDL are new and not fully explored things for me, so I hope to learn them better with your case.
I do not know if you stopped by our office hours yesterday, as I got involved in helping another user. I would be happy to meet with you sometime this week to make sure everything works.
Thank you! When I downloaded containers I had not set singularity cache first. I did what you suggested and confirmed the container images where in the folder singularity-cache/pull
Unfortunately it failed, Im not sure it was able to pick the container up from cache and job failed as reported in log (edited user)
/scratch1/trojan/cromwell/cromwell-executions/MultiSampleSmartSeq2/910a4b60-3a32-4ce8-a4d5-586dd84ae5fa/call-sc_se/shard-0/SmartSeq2SingleCell/cedd7049-6203-4a0a-99fd-31f77f5ac634/call-HISAT2SingleEndTranscriptome/execution/script.submit: line 5: /scratch1/trojan/cromwell/set_singularity_cachedir.sh: No such file or directory
mkdir: cannot create directory '/scratch1/trojan': Permission denied
WARNING: Cache disabled - cache location /scratch1 is not writable.
INFO: Starting build...
FATAL: While performing build: conveyor failed to get: pinging container registry quay.io: Get "https://quay.io/v2/": dial tcp: lookup quay.io on [::1]:53: read udp [::1]:43966->[::1]:53: read: connection refused
sbatch: error: invalid partition specified: (null)
sbatch: error: Batch job submission failed: Invalid partition name specified
I do have the file /scratch1/trojan/cromwell/set_singularity_cachedir.sh.
Thank you for helping me on this and happy to meet. Let me know when and how. I am available this afternoon and tomorrow afternoon.
Please, replace trojan with your username rklotz in the Cromwell config file.
I must have missed that information before
Oh yes it is with my username. I thought i would edit it for this forum conversation
please check again lines 208 and 209 of the cromwellslurmsingularity.conf for the username. It might be misspelled
My mistake! the username was misspelled! I feel we are close. Still isn’t working tho:
INFO: Starting build...
FATAL: While performing build: conveyor failed to get: pinging container registry registry-1.docker.io: Get "https://registry-1.docker.io/v2/": dial tcp: lookup registry-1.docker.io on [::1]:53: read udp [::1]:41013->[::1]:53: read: connection refused
sbatch: error: invalid partition specified: (null)
sbatch: error: Batch job submission failed: Invalid partition name specified
INFO: Starting build...
FATAL: While performing build: conveyor failed to get: pinging container registry quay.io: Get "https://quay.io/v2/": dial tcp: lookup quay.io on [::1]:53: read udp [::1]:46006->[::1]:53: read: connection refused
sbatch: error: invalid partition specified: (null)
sbatch: error: Batch job submission failed: Invalid partition name specified
Here is an overview of singularity cache in case it is usefull:
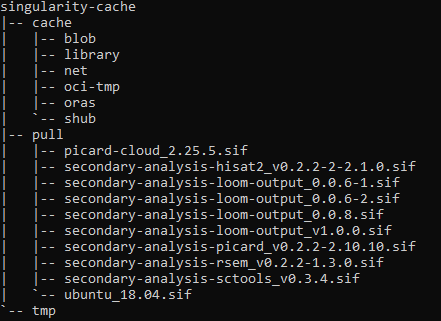
During workflow I can see this
# Build the Docker image into a singularity image
IMAGE=quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0.sif
or
--wrap "singularity exec --bind /scratch1/rklotz/cromwell/cromwell-executions/MultiSampleSmartSeq2/278c5068-2b50-4bd3-9c4b-af988e2e4d04/call-sc_se/shard-1/SmartSeq2SingleCell/9d9ddfb7-0cf2-4c2b-84ee-217eefd2da14/call-HISAT2SingleEnd:/cromwell-executions/MultiSampleSmartSeq2/278c5068-2b50-4bd3-9c4b-af988e2e4d04/call-sc_se/shard-1/SmartSeq2SingleCell/9d9ddfb7-0cf2-4c2b-84ee-217eefd2da14/call-HISAT2SingleEnd quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0.sif /bin/bash /cromwell-executions/MultiSampleSmartSeq2/278c5068-2b50-4bd3-9c4b-af988e2e4d04/call-sc_se/shard-1/SmartSeq2SingleCell/9d9ddfb7-0cf2-4c2b-84ee-217eefd2da14/call-HISAT2SingleEnd/execution/script"
It calls for quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0.sif when the image in cache is saved as secondary-analysis-hisat2_v0.2.2-2-2.1.0.sif. Could be the reason why workflow is unable to start job?
This is the moment when I started having issues.
I did some testing and please edit cromwellslurmsingularity.conf file:
- line 233 - please comment that line out
- line 234 - please comment that line out
- line 248 - please change
${docker}.sifto${docker}
Pulling the docker image with singularity should do everything necessary and place the image in the cache directory
This should take care of the container image
line 233-234
IMAGE=${docker}.sif
singularity build $IMAGE docker://${docker}
Changing line 248 does not resolve problem
What is the error message?
changing line 248 ${docker}.sif to ${docker} produces these errors
INFO: Starting build...
FATAL: While performing build: conveyor failed to get: pinging container registry registry-1.docker.io: Get "https://registry-1.docker.io/v2/": dial tcp: lookup registry-1.docker.io on [::1]:53: read udp [::1]:39520->[::1]:53: read: connection refused
sbatch: error: invalid partition specified: (null)
sbatch: error: Batch job submission failed: Invalid partition name specified
INFO: Starting build...
FATAL: While performing build: conveyor failed to get: pinging container registry quay.io: Get "https://quay.io/v2/": dial tcp: lookup quay.io on [::1]:53: read udp [::1]:49284->[::1]:53: read: connection refused
sbatch: error: invalid partition specified: (null)
sbatch: error: Batch job submission failed: Invalid partition name specified
Same if I also change line 233 IMAGE=${docker}.sif by IMAGE=${docker}
That is the correct error message. Have you specified the partition name in your WDL/json file in the section runtime? cromwell config needs to know that information. Please add the partition and account name to your runtime section in WDL (of course edit to reflect the correct names). I think you are getting really close
String partition
String account
runtime {
partition: "${partition}"
account: "${account}"
docker: docker
memory: "${machine_mem_mb} MiB"
disks: "local-disk ${disk} HDD"
cpu: cpu
preemptible: preemptible
and to the json file:
runtime {
partition: "main"
account: "rklotz_600"
docker: "ubuntu:18.04"
cpu: 1
memory: "1 GiB"
disks: "local-disk 1 HDD"
}
I tried specifying account and partition in WDL files. Here is the error now
INFO: Starting build...
FATAL: While performing build: conveyor failed to get: pinging container registry quay.io: Get "https://quay.io/v2/": dial tcp: lookup quay.io on [::1]:53: read udp [::1]:48491->[::1]:53: read: connection refused
I tried adding partition and account in json file but the input workflow failed for unexpected input provided
It looks like singularity tries to build the requested container.
Are the changes below applied to the cromwell config file?
- line 233 - please comment that line out
- line 234 - please comment that line out
- line 248 - please change
${docker}.sifto${docker}
If I do that, it still fails. But what’s weird is that the stderr.submit log is empty. When workflow failed I see this
--wrap "singularity exec --bind /scratch1/rklotz/cromwell/cromwell-executions/MultiSampleSmartSeq2/0e02cdef-f505-40ef-a0da-423537054e8b/call-sc_se/shard-0/SmartSeq2SingleCell/cee9a69f-e1af-4802-863e-974a5bc0c0ff/call-HISAT2SingleEnd:/cromwell-executions/MultiSampleSmartSeq2/0e02cdef-f505-40ef-a0da-423537054e8b/call-sc_se/shard-0/SmartSeq2SingleCell/cee9a69f-e1af-4802-863e-974a5bc0c0ff/call-HISAT2SingleEnd quay.io/humancellatlas/secondary-analysis-hisat2:v0.2.2-2-2.1.0 /bin/bash /cromwell-executions/MultiSampleSmartSeq2/0e02cdef-f505-40ef-a0da-423537054e8b/call-sc_se/shard-0/SmartSeq2SingleCell/cee9a69f-e1af-4802-863e-974a5bc0c0ff/call-HISAT2SingleEnd/execution/script"
[2022-04-14 14:08:40,28] [info] WorkflowManagerActor: Workflow 0e02cdef-f505-40ef-a0da-423537054e8b failed (during ExecutingWorkflowState): java.lang.RuntimeException: Unable to start job. Check the stderr file for possible errors: /scratch1/rklotz/cromwell/cromwell-executions/MultiSampleSmartSeq2/0e02cdef-f505-40ef-a0da-423537054e8b/call-checkArrays/execution/stderr.submit
Before I apply changes in line 233, 234 and 248 the stderr file shows
INFO: Starting build...
FATAL: While performing build: conveyor failed to get: pinging container registry quay.io: Get "https://quay.io/v2/": dial tcp: lookup quay.io on [::1]:53: read udp [::1]:48491->[::1]:53: read: connection refused
After editing line 233, 234 and 248 stderr file is empty