I’m trying to use the Bioperl Bio::DB::Taxonomy module but am getting the following @INC error when loading the available CARC modules using:
module purge
module load gcc perl perl-bioperl
I tried a local install using cpanm:
cpanm -l /project/ckenkel_26/software/perl_mods Bio::DB::Taxonomy
including adding the following line to my perl script
use local::lib ‘/project/ckenkel_26/software/perl_mods’;
But this throws another @INC error for local::lib
Some progress, but still having an install issue - the @INC error is now ‘Can’t locate DB_File.pm’ so I’ve tried to install this using cpanm but the install fails with an ‘unable to locate db.h’
I searched online and it appears that this error can arise when Berkeley DB is in a nonstandard location. See README - metacpan.org
I tried to do a source install for DB_File.pm pointing to the berkeley-db/18.1.40 module /include directory but then it tries to install it centrally but I don’t have permission to write to the perl 5.36.0 module directory and I can’t see a way to run a local install when building from the source code
another issue is that to use the Berkeley DB module (berkeley-db/18.1.40) I need gcc/11+ but the version of perl-bioperl installed requires gcc/8.3.0 which I fear will lead to some additional incompatibility issues…although perhaps Berkeley isn’t needed once the DB_File.pm install is complete…
It may be easier to use a container in this case. I see an existing Docker image here, though it’s a few years old: Docker
The image seems to have all of the BioPerl modules already installed. Do you want to try that out? You would need to convert the image to the Singularity format in order to run on CARC systems:
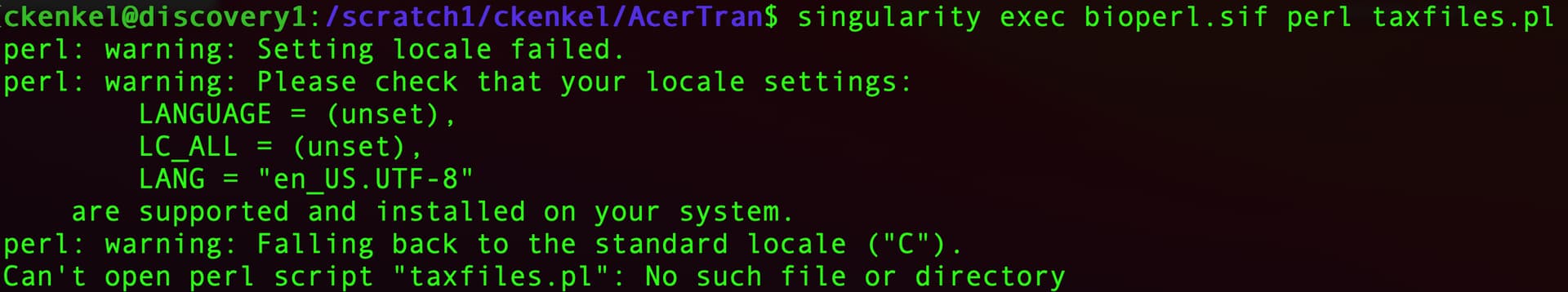
Okay - that worked and the build finished with no errors that I could see. But when I try to run my script I get weird path warnings - is there some other flag I need to add to get it to register that all my files are in the directory I’m working in? For example - I did a simple test with perl -v and it returns this error but then prints a version of perl (which is different than the standard default one on the login node).
good news - this solution worked for this particular script and I was able to submit it as a job!
bad news - I have a second script requiring bioperl that includes a system call for a blast command - blastdbcmd - and this does not work, presumably because the container is bioperl only. Your video training makes it pretty clear that containers are unique and can’t be modified, but I figured it would be worth a final ask - is there any way to add to an existing container?
Yes, you could build a Singularity container image using this container image as the base image in the definition file and then install more software over that.
We have Blast+ installed as a module, so you may be able to bind /spack to the container and then provide the path to the binary: