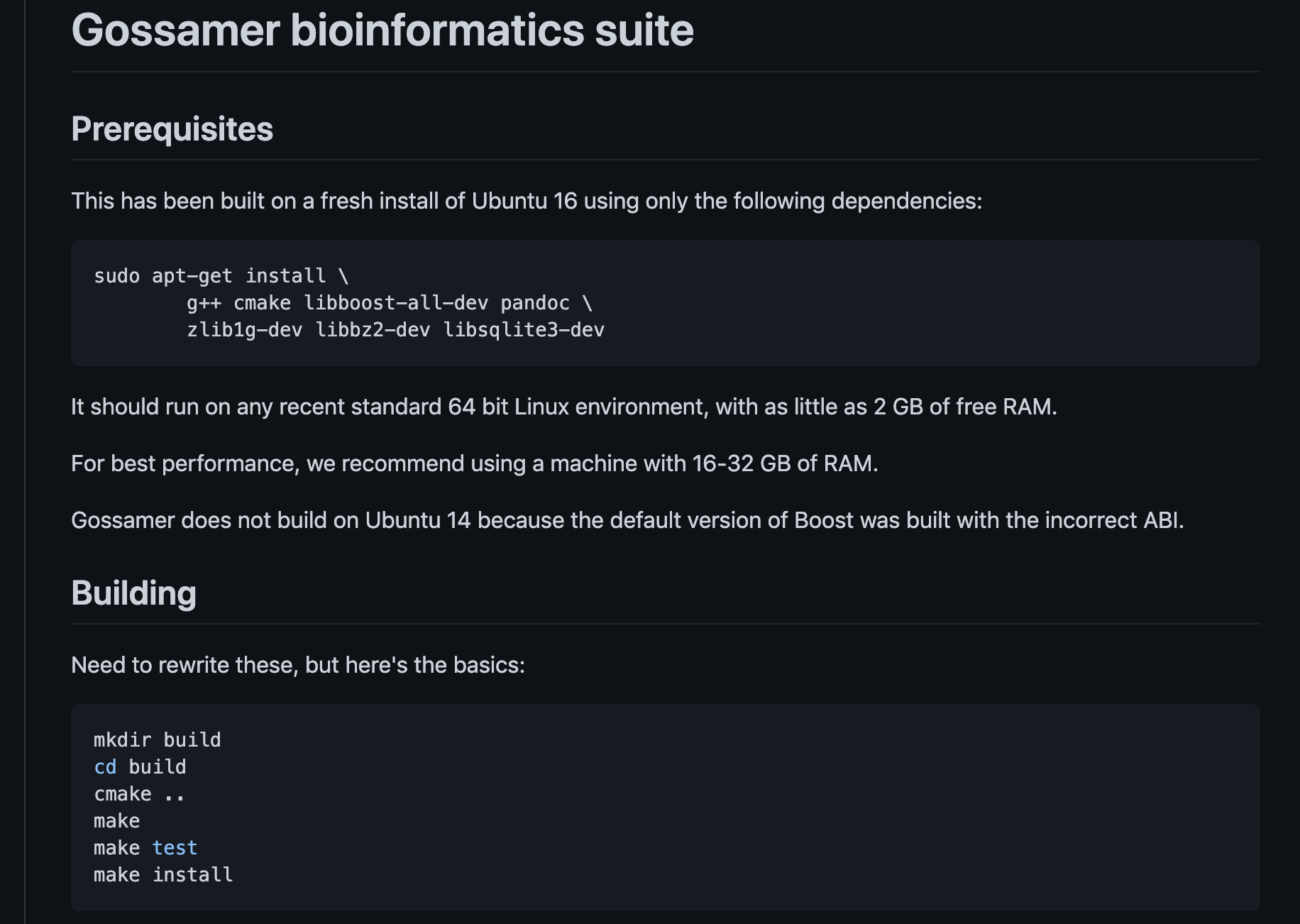
Hi I am trying to install the tool as in the below instruction but I could not on discovery, would you please provide some guides?
Thank you.
Mohamed
Hi I am trying to install the tool as in the below instruction but I could not on discovery, would you please provide some guides?
Thank you.
Mohamed
@salehm It seems the source code would need to be modified. Instead, I found a Docker container image that you could convert to a Singularity format in order to use on CARC systems: https://hub.docker.com/r/zlskidmore/gossamer
We have a guide for Singularity here: https://carc.usc.edu/user-information/user-guides/software-and-programming/singularity
@dstrong Thank you so much for the instructions you provided, very much useful. However, when I tried to pull the image it gave a fatal error
“FATAL: While making image from oci registry: error fetching image to cache: while building SIF from layers: packer failed to pack: while unpacking tmpfs: error unpacking rootfs: unpack layer: unpack entry: usr/lib/python3.7/distutils/README: link: unpriv.link: unpriv.wrap target: operation not permitted”
I loaded python but it still giving the same error.
Your support is very much appreciated.
Thanks.
Mohamed
Conversions from some Docker containers do not work with the pull command; I’m not sure exactly what the reason is. It should work if you try the remote builder with a simple definition file:
Bootstrap: docker
From: zlskidmore/gossamer
@salehm I did some testing and it seems the pull command fails if the TMPDIR or SINGULARITY_TMPDIR variables are set to a directory on one of the file systems. You could try entering unset TMPDIR, which will then revert to using the local /tmp space on the login node, and then try pulling again. This is working for me.
@dstrong Thank you Derek, just saw your last message. I already created the definition file and pulled the image. However, when I tried to execute a command it would not work.
Please find the attached screenshot.
Thanks.
Mohamed
You can run the singularity exec command directly; there’s no need to run singularity shell first. To exit the Singularity container shell, type exit.
@dstrong Thank you. When I did that, the command I am running to build a genome index using human and mouse genomes, could find the human genome file. Any idea why this could be?
I re downloaded both the human and mouse genomes and put them all together with the image.sif file in one directory but still could not find the human file.
Thanks for your support.
Mohamed
Are these files all in your home directory or another directory? If in another, you would need to --bind that directory to the container. For example:
singularity exec --cleanenv --bind $PWD image_latest.sif xenome ...
@dstrong you right the files were in the proj diirctory not the home directory. I tried binding the directory the container software but also would not work. I then set the environment by adding "export singularity_bind=<path_to_direc> in the .bash_profile but it also did not work.
Only when I copied and pasted the files to the home directory I did not get the error message but it still running. The command should take 6-8 hours to finish. Hopefully it works okay.
Thank you for your support all the way long, and I will keep you updated.
Mohamed
@dstrong
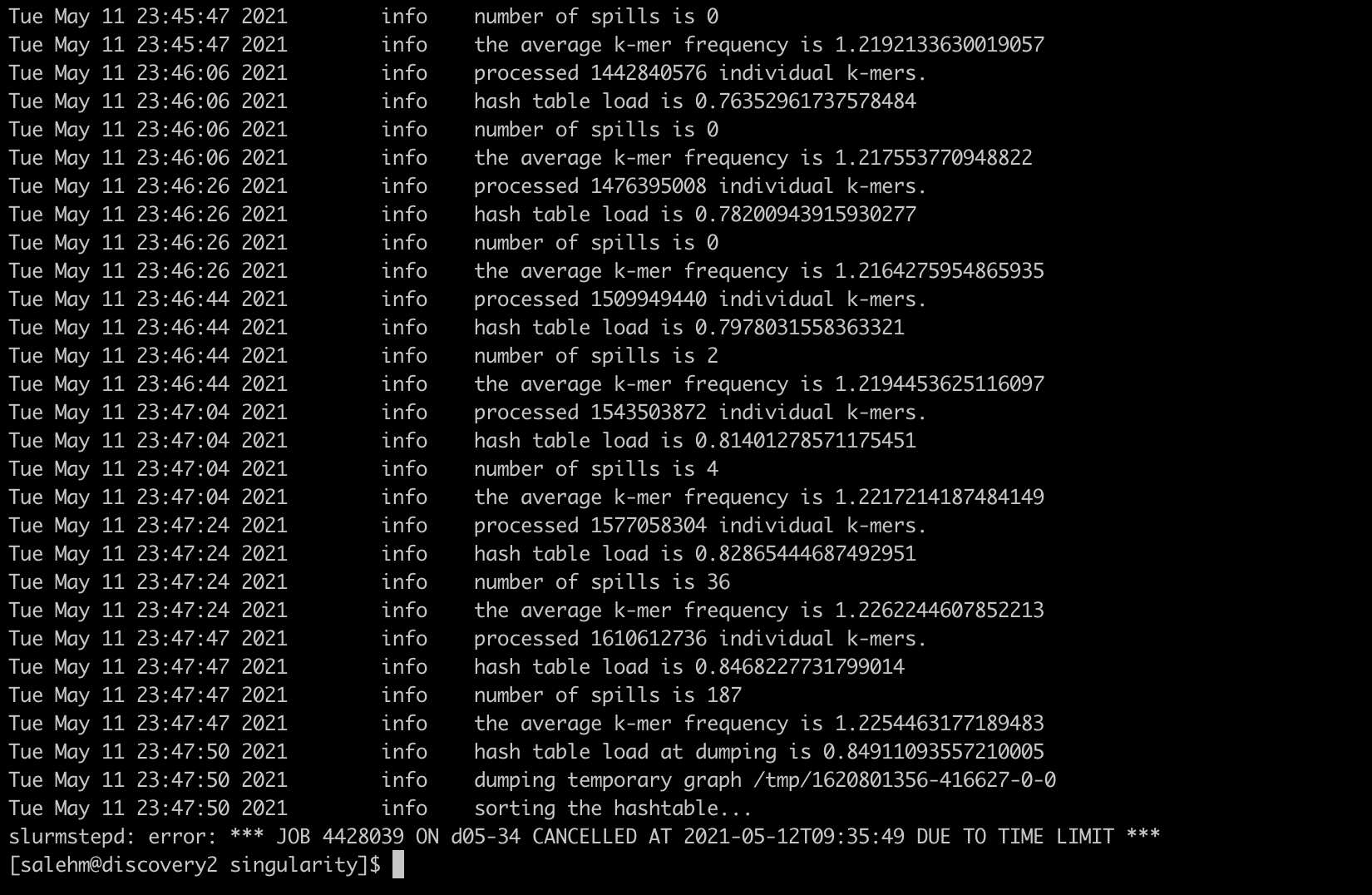
Although I gave it 10h:30min to run, xenome failed for time limit out. I tried again with 24hours but it also failed. Please see the screenshot of my slurm order. The developer says it should take 6-8 hours for indexing. I am not sure why its taking too long. In the meantime I submitted a slurm job with 96 hours and another one (for small genomes as a quick test) for 48:30, however both job are pending for sometimes now.
Your suggestions are very welcome.
Thank you for your help.
Mohamed
The job statistics show that the cores are not being fully utilized and memory use is maxed out. Does the Slurm .out file provide any relevant information?
Try a job script like the following:
#!/bin/bash
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem=32GB
#SBATCH --time=10:00:00
module purge
singularity exec image_latest.sif xenome index -v -M 32 -T 8 -P idx -H mouse.fa -G human.fa
@dstrong
Thank you for the script. I tried it but unfortunately it still giving the titmeout message. Please find below a screenshot of the end of the slurm.out file.
Should I increase the time and try again?
Thanks for your patience.
Mohamed
@dstrong I found this conversation on the package webpage
Maybe if I increase RAM to 96G it may help.
I am actually not sure if the package is not compatible with our server?
Thanks.
Mohamed
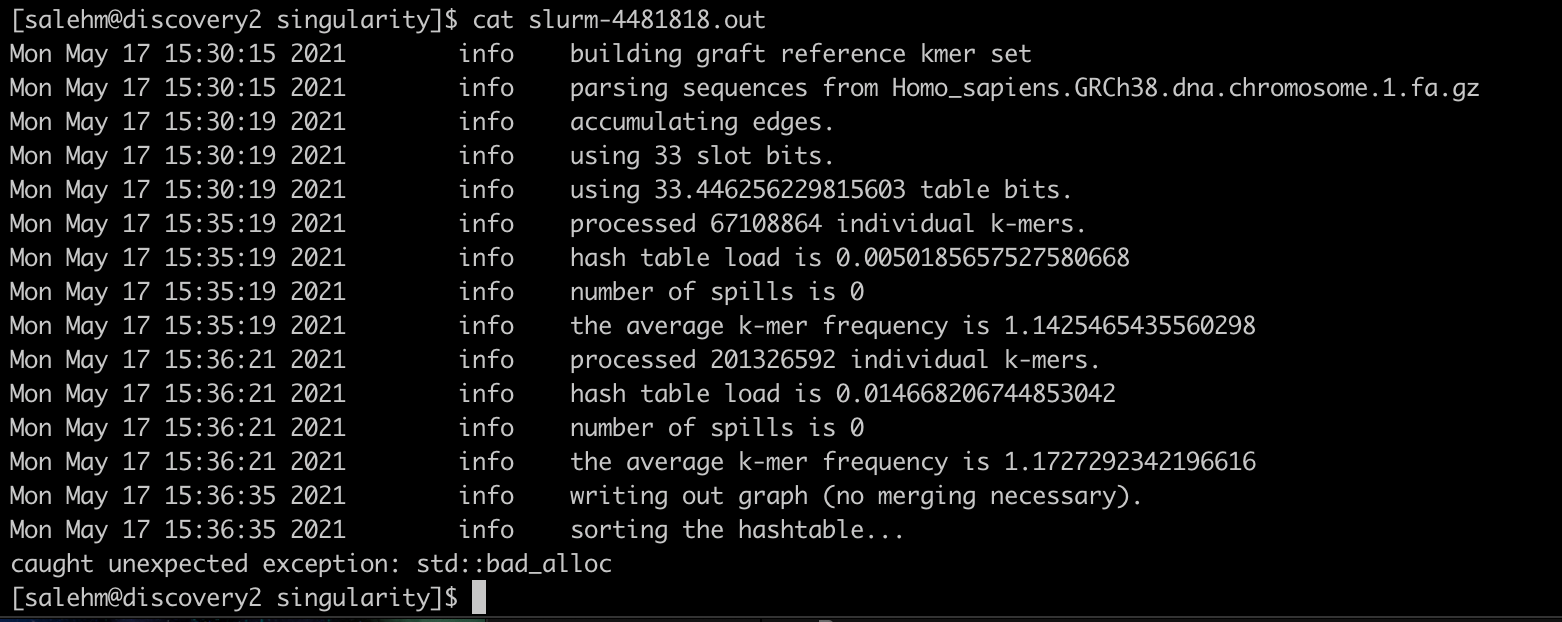
@dstrong
Based on the above conversation, I tried 96G RAM, which seemed worked much faster but it failed due to the attached error. Shall I increase the memory to 120 G for example?
Thanks.
Mohamed
It’s the same issue with the cores not being fully utilized, appears to only be using 1 core, which I assume is why it’s taking longer than expected. Not sure why that’s happening. The documentation states that “The actual number of threads used during the algorithms depends on each implementation.” but offers nothing more than that. Unfortunately the development of this software stopped 4 years ago.
If increasing memory helps, try a job script like the following:
#!/bin/bash
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mem=120GB
#SBATCH --time=48:00:00
#SBATCH --partition=oneweek
module purge
singularity exec image_latest.sif xenome index -v -M 120 -T 8 -P idx -H mouse.fa -G human.fa
On the oneweek partition you could increase the memory request up to a max of 248 GB.
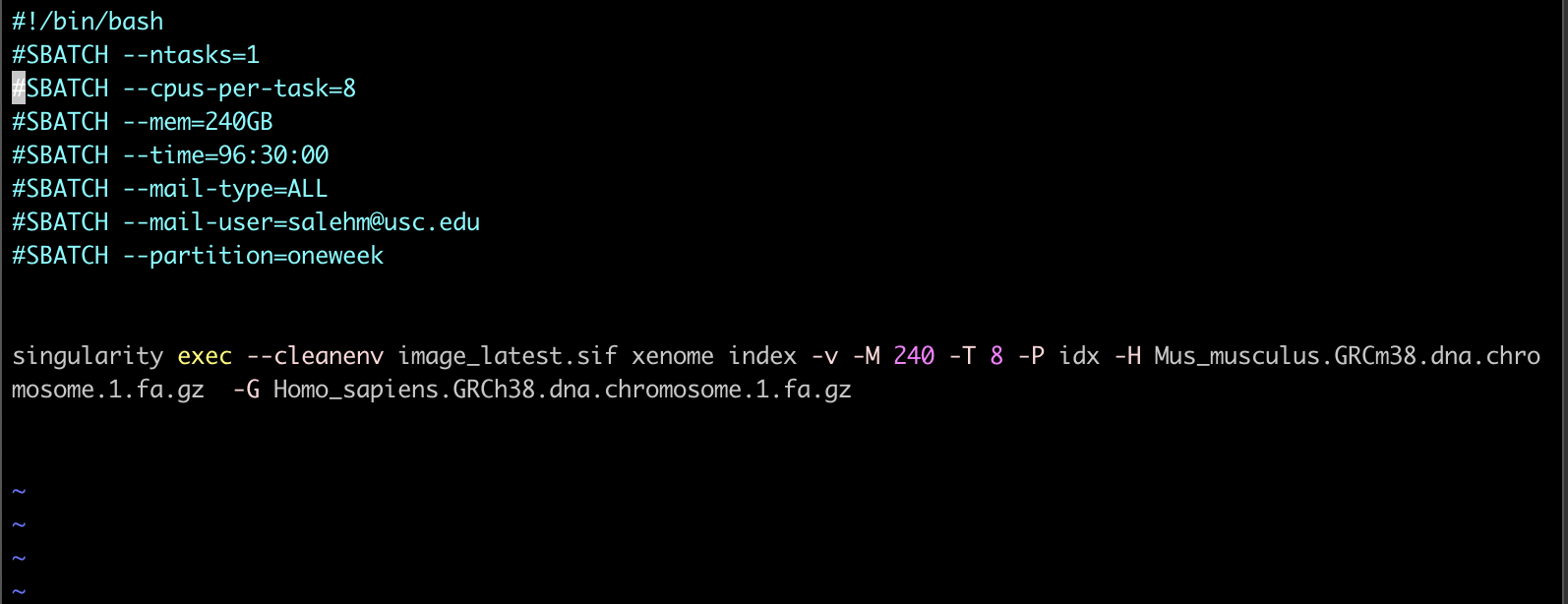
@dstrong
I tried again with more memory but this time using only one chromosome of each genome (so much smaller files) and gave me the error below. Note, it ran only for 4 hours then failed. Please also see my job script. I think if I request the right resources, I may get it to work. Would you advice on that?
Thanks.
It would appear the job ran out of memory, although the job statistics show it still had about 18 GB free. Try increasing the request to 248 GB and 16 CPUs, but only give xenome about 246 GB to preserve some memory for overhead.